Корзина пуста

rhVEGF-A121 фактор роста эндотелия сосудов- А человека, изоформа 121, рекомбинантный белок

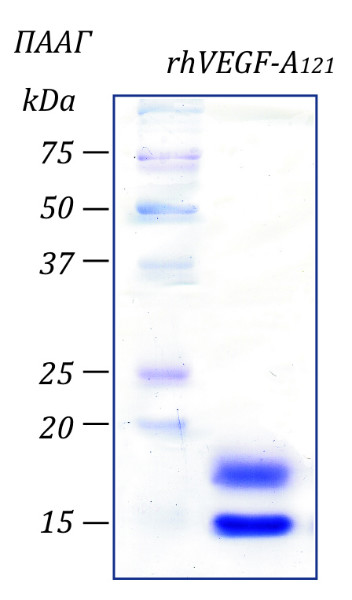
Источник: Клеточная линия CHO, продуцирующая rhVEGF-A121.
Чистота: >98% в соответствии с электрофорезом в ПААГ с последующим окрашиванием Coomassie Brilliant Blue.
Уровень эндотоксина: <1.0 EU на 1 мкг белка, LAL-тест.
Структура: Дисульфид-связанный гомодимер.
Молекулярный вес: В ПААГ:15-19 кДа в редуцирующих условиях
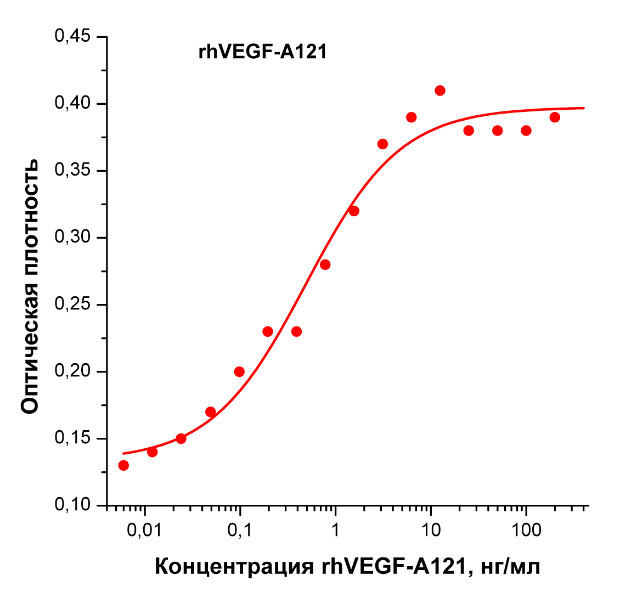
Биологическая активность: Рекомбинантный белок rhVEGF-A121 человека стимулирует пролиферацию клеток эндотелия пупочной вены человека (HUVEC). ED50 для данного эффекта обычно 0,3 - 1,5 нг/мл. Оптимальная концентрация для индивидуального применения определятся пользователем.
Форма
Лиофильно высушен из фосфатного буферного раствора PBS, содержащего 0,05% Tween20, pH 7.0, профильтрованного через фильтр с диаметром пор 0,22мкм. Не содержит вспомогательных белков.
Разведение
Центрифугировать флакон при 1000g, 3 мин. Добавить стерильный фосфатный буферный раствор (PBS) до конечной концентрации 0,1-1 мкг/мкл. Оставить на 20-30 мин при комнатной температуре, затем центрифугировать при 1000g в течение 1 мин, и мягко ресуспендировать. Для приготовления рабочих растворов можно использовать буфер на водной основе или культуральную среду. Добавление вспомогательных белков (BSA или FBS) не является необходимым.
Условия транспортировки
Перевозить при комнатной температуре.
Стабильность и Условия хранения:
- 24 месяца, хранение невскрытой упаковки, при температуре от -20 до -70°C.
- 1 месяц, разведенный в стерильных условиях, при температуре от 2 до 8°C.
- 6 месяцев, разведенный в стерильных условиях, при температуре от -20 до -70°C.
Не рекомендуются повторные циклы замораживания-оттаивания раствора рекомбинантного белка.
| Описание | |
| Дополнительное описание и цитирование | Фактор Роста Эндотелия Сосудов (VEGF, от англ. Vascular endothelial growth factor) является одним из ключевых регуляторов ангиогенеза. Наиболее важную роль в организме человека играет белок семейства VEGF, называемый VEGF-A. В данное семейство также входят плацентарный фактор роста (PlGF) и белки VEGF-B, VEGF-C, VEGF-D. VEGF-A представляет собой гликопротеиновый гомодимер, 45кДа, и играет ключевую роль в хемотаксисе и дифференцировке ангиобластов, в пролиферации эндотелиоцитов (ЭК), в васкулогенезе и в перестройке сосудов. В результате альтернативного сплайсинга или протеолитического расщепления формируются 4 основные изоформы VEGF-A, состоящие из 121, 165, 189 или 206-ти аминокислотных остатков. Достоверно установленным действием VEGF является ускорение роста эндотелиоцитов артерий, вен и лимфатических сосудов. VEGF индуцирует мощный ангиогенный ответ во множестве моделей in vivo. Кроме того, VEGF увеличивает проницаемость сосудов, и это свойство лежит в основе важной роли молекулы в воспалении и других патологических процессах. В данном контексте VEGF также индуцирует экспрессию эндотелием некоторых молекул адгезии, регулирующих адгезию лейкоцитов при воспалении. In vitro VEGF предотвращает апоптоз эндотелиоцитов, индуцируемый истощением сыворотки. VEGF также индуцирует экспрессию в эндотелиоцитах антиапоптотического белка BCL2. Зависимость от VEGF была показана на эндотелиоцитах вновь образованных, но незрелых сосудов опухолей. Следует подчеркнуть, что хотя эндотелиальные клетки являются главными мишенями VEGF, в ходе нескольких исследований были показаны также его эффекты на митотическую активность/выживаемость некоторых неэндотелиальных клеточных типов, включая нейроны и опухолевые клетки. VEGFs-сигнализация осуществляется через рецепторные тирозинкиназы VEGFR, наиболее значимой из которых является VEGFR-2 (Flk-1/KDR). VEGF-A, связываясь с VEGFR-2, вызывает димеризацию и аутофосфорилирование тирозинкиназных остатков. Активация фосфолипазы С и последующий запуск MAPK-киназного каскада лежит в основе формирования новых сосудов. VEGFR-2 стимулирует фосфорилирование T-специфического белка-адаптора, который ассоциирован с цитоплазматической тирозинкиназой Src, регулирующей организацию актиновых фибрилл и миграцию эндотелиальных клеток в ответ на VEGF-A. Подобным образом, VEGF-A через активацию Src-зависимой малой ГТФазы Rac обеспечивает зависимое от р21-киназы фосфорилирование высококонсервативных тирозиновых остатков VE-кадгерина, что приводит к его интернализации и разъединению ЭК, ведущему к увеличению проницаемость эндотелия. Кроме того, связывание VEGF-A с VEGFR-2 активирует путь фосфоинозитид-3-киназы (PI3K) с последующим увеличением уровня экспрессии антиапоптотических белков, например, Bcl-2, и снижением активности проапоптотических белков, таких, как каспаза-3. Нарушение баланса между стимуляторами и ингибиторами роста сосудов и, как следствие, чрезмерный или недостаточный ангиогенез, играет серьёзную роль в патогенезе многочисленных заболеваний. Избыточный ангиогенез сопровождает злокачественные опухоли, воспалительные заболевания и поражения глаз. Вносит свой вклад в течение таких патологий, как астма, цирроз, эндометриоз, ожирение, СПИД, диабет, аутоиммунные заболевания и т.д. Ряд других патологий - ишемия сердца, инсульт, язвенные поражения, нейродегенерации - характеризуется недостаточным ангиогенезом, слабым снабжением поражённых тканей кровью. Понимание основных механизмов ангиогенеза в норме и его нарушений при конкретных болезнях лежит в основе выработки успешной терапии различных заболеваниий. Использованная литература:
|